ARG1-D jest odrębnym zaburzeniem cyklu mocznikowego (UCD)8.
ARG1-D, również znana jako hiperargininemia, jest autosomalną recesywną chorobą spowodowaną przez mutacje w genie ARG1, który koduje enzym arginazę 1 (ARG1)2,9.
Enzym ARG1 rozkłada argininę na ornitynę i mocznik w ostatnim etapie cyklu mocznikowego. Cykl mocznikowy obejmujący pięć głównych etapów odbywa się głównie w wątrobie i przekształca amoniak (NH3), pochodzący z rozpadu białek, w mocznik10.
- Arginina to aminokwas, który odgrywa ważną role w utrzymywaniu homeostazy naczyniowej oraz innych funkcji fizjologicznych11.
- W przypadku braku funkcjonalnego enzymu, arginina i metabolity związane z argininą, w tym amoniak i związki guanidynowe, gromadzą się i są związane z patologiami neuromotorycznymi12,13.
- Trwale podwyższony poziom argininy u pacjentów z ARG1-D jest kluczowym czynnikiem wywołującym objawy choroby takie jak: postępująca spastyczność, opóźnienia rozwojowe oraz drgawki4,5,14,15.
- Hiperamonemia nie jest objawem patognomicznym ARG1-D a ostre epizody hiperamonemii występują rzadko14-16.
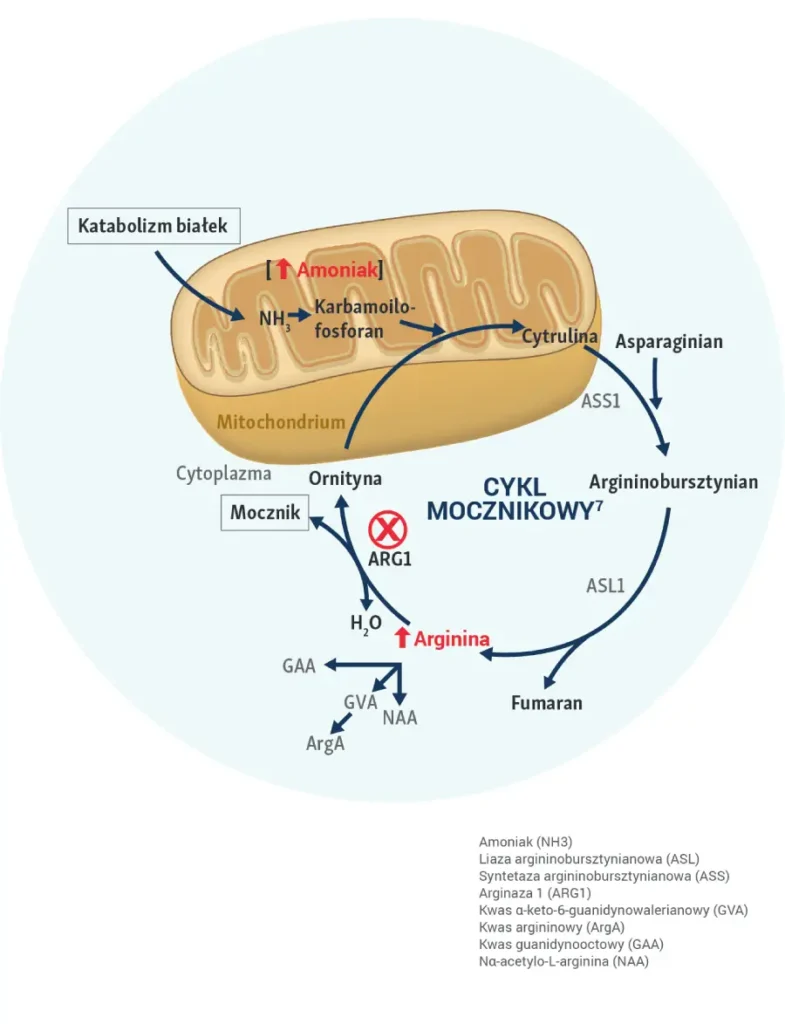
Stale podwyższony poziom argininy i jej metabolitów w osoczu naraża pacjentów na zwiększone ryzyko zachorowalności i wczesnej umieralności1-7.
Dotyka dzieci i trwa do dorosłości, pacjenci prezentują heterogeniczne formy ARG1-D2,5,18-20.
Kluczowymi objawami choroby, które mogą wystąpić, są: postępująca spastyczność, opóźnienie rozwojowe, upośledzenie intelektualne oraz drgawki2,5,19.
- Objawy zwykle pojawiają się w dzieciństwie i utrzymują się z czasem, ze względu na kumulację argininy w osoczu2.
- Spastyczność zwykle ujawnia się po kilku pierwszych miesiącach życia i stopniowo nasila się w wieku dorosłym2,9,12,16.
- Diplegia spastyczna oraz chód na palcach są częstymi objawami ARG1-D2,16.
- Początek objawów jest zwykle ograniczony do kończyn dolnych; jednak w miarę nasilania się spastyczności może dojść również do porażenia kończyn górnych2.
- Pacjenci wykazują postępujące i zmienne pogorszenie funkcji neurologicznych i rozwojowych oraz umiejętności funkcjonalnych2,5,19,21.
Wysokie stężenia argininy w osoczu ostatecznie odróżniają ARG1-D od innych zaburzeń cyklu mocznikowego, jak również od chorób neurometabolicznych i neurologicznych takich jak porażenie mózgowe lub dziedziczna paraplegia spastyczna2,4,12.
Referencje:
1. Diez-Fernandez C, et al. Hum Mutat. 2018;39:1029-1050. 2. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 3. Häberle J, et al. J Inherit Metab Dis. 2019;1–39. 4. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 5. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 6. Sun A, et al. Arginase deficiency. In: Adams MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 7. Diaz GA, et al. Poster presented at: 13th European Paediatric Neurology Society (EPNS) Congress; September 17-21, 2019; Athens, Greece. Poster P06-34. 8. Carvalho DR, et al. Gene. 2012;509:124-130. 9. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 10. Barmore W, et al. Physiology, Urea Cycle. [Updated 2021 May 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan. Available at: https://www.ncbi.nlm.nih.gov/books/NBK513323. Accessed December 3, 2021. 11. Tapiero H, et al. Biomed Pharmacother. 2002;56:439-445. 12. Prasad A, et al. J Child Neurol. 1997;12:301-309. 13. Amayreh W, et al. Dev Med Child Neurol. 2014;56:1021-1024. 14. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 15. NORD. The Physician’s Guide to Urea Cycle Disorders. 2012. Available at: http://www.nucdf.org/documents/NORD_Physician_Guide_to_Urea_Cycle_Disorders.pdf. Accessed November 26, 2021. 16. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 17. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 18. Sin YY, et al. J Mol Med (Berl). 2015;93:1287-1296. 19. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 20. Bakhiet M, et al. Medicine (Baltimore). 2018;97:e10780. 21. Schlune A, et al. Amino Acids. 2015;47:1751-1762.