Objawy ARG1-D często przypominają objawy innych neurologicznych i neurometabolicznych chorób, takich jak inne zaburzenia cyklu mocznikowego, porażenie mózgowe lub dziedziczna paraplegia spastyczna5,6.
Diagnostyka różnicowa ARG1-D polega na zidentyfikowaniu objawów klinicznych związanych z wysokim stężeniem argininy w osoczu4-7.
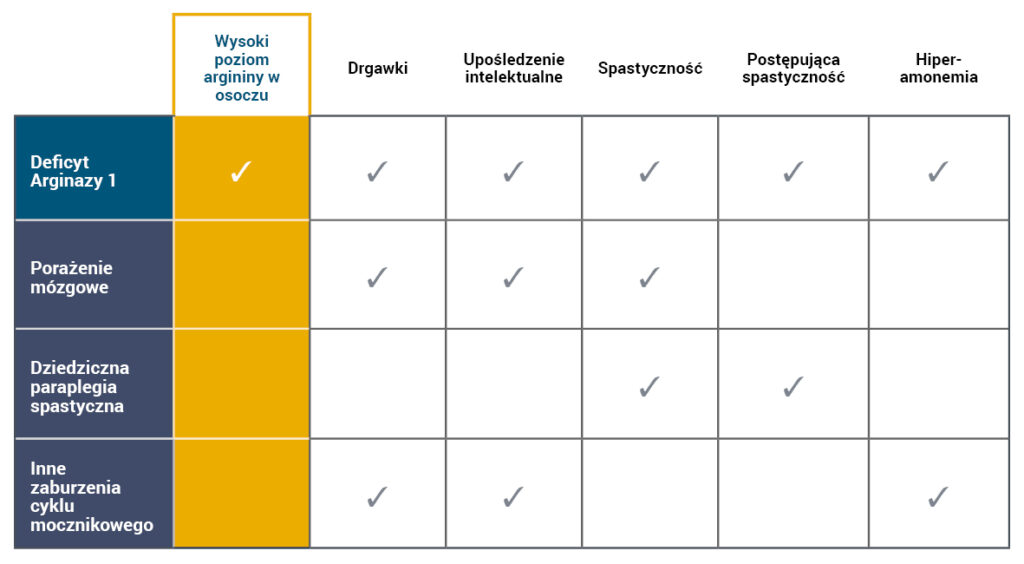
- Hiperamonemia nie jest objawem patognomicznym ARG1-D a ostre epizody hiperamonemii występują rzadko4,8.
Z powodu ograniczeń badań przesiewowych noworodków, ARG1-D może być pominięty z wielu powodów:
- Określenie wartości normy dla poziomu argininy w badaniach przesiewowych jest problematyczne, ponieważ przenoszenie metabolitów, takich jak arginina, z matki na dziecko może utrudniać badania9,10.
- Algorytmy przesiewowe i normy poziomu argininy różnią się9.
- ARG1-D nie jest uwzględniony w badaniach przesiewowych noworodków w większości krajów europejskich11.
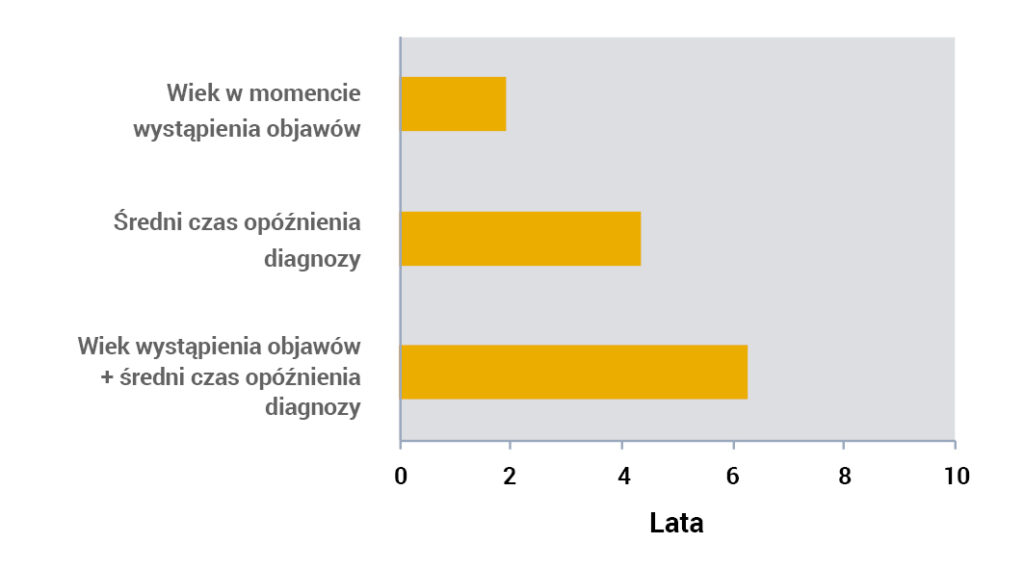
Opóźnienia w diagnozie w połączeniu z późnym wystąpieniem objawów prowadzi do podjęcia pierwszej interwencji dopiero w wieku około 6 lat1.
Rutynowe badanie panelu aminokwasów w osoczu połączone z badaniami genetycznymi może potwierdzić ARG1-D12,13.
Przed postawieniem wstępnego rozpoznania ważne jest przeprowadzenie pełnego wywiadu medycznego, dietetycznego, rodzinnego i społecznego oraz wykonanie dokładnego badania fizycznego.
Sprawdź wysoki poziom argininy, który powoduje objawy ARG1-D, rutynowymi testami3,12,13.


Jeśli występują wysokie poziomy argininy w osoczu, badania genetyczne† mogą potwierdzić diagnozę.
†Ze względu na heterogenność genotypów ARG1, nie wszystkie mutacje powodujące ARG1-D zostały zidentyfikowane.
Referencje:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.